CORE SERVICES
Overview

A brief overview of core services:
• Single- and paired-end sequencing
• Multiplexing
• Library construction services
• Library quality control
• Data analysis services
NextGen Sequencing
 Up to 8 samples may be run on a single flow cell, or multiple samples may be multiplexed in one lane if obtaining the absolute highest coverage is not necessary or if previous runs have yielded excessive coverage (this may reduce costs). When deciding, keep in mind that our HiSeq is currently putting out 150+ million reads. Our core is actively engaged with Illumina scientists and other NextGen sequencing leaders to ensure we are up-to-date with reagents, software, and parts, and quality is constantly monitored throughout the run.
Up to 8 samples may be run on a single flow cell, or multiple samples may be multiplexed in one lane if obtaining the absolute highest coverage is not necessary or if previous runs have yielded excessive coverage (this may reduce costs). When deciding, keep in mind that our HiSeq is currently putting out 150+ million reads. Our core is actively engaged with Illumina scientists and other NextGen sequencing leaders to ensure we are up-to-date with reagents, software, and parts, and quality is constantly monitored throughout the run.
We currently offer:
• 50 cycle single read or paired end reads
• 100 cycle single read or paired end reads
• Multiplexing for any read type
For recommendations about which type of run best suits your needs, please contact us.
Library Validation
We strongly recommend that all libraries are validated and quantitated using Bioanalyzer, qPCR, or similar methods before submission. In addition, the core staff validates all submitted libraries before sequencing to ensure maximum quality and cluster yield. This service is included in the sequencing cost. Please click the "library validation" box in Galaxy when submitting your samples if you are submitting barcoded subsamples and need us to pool them for you.
Library Construction
Many researchers prepare their own libraries using unique or experimental protocols. Alternatively, the core facility can prepare the libraries for you. The core can work with you to identify or devise a library construction method that fits your needs. The lab is prepared to create genomic and ChIP-seq libraries, and is gearing up to offer mRNA, sRNA, strand-specific mRNA, as well as some target enrichment library construction strategies, such as the Agilent SureSelect system (bait sequences not supplied).
To discuss your library construction options and pricing, please contact us, or click here for basic pricing information.
To see a list of established library construction protocols and other useful information, click here.
Data Analysis
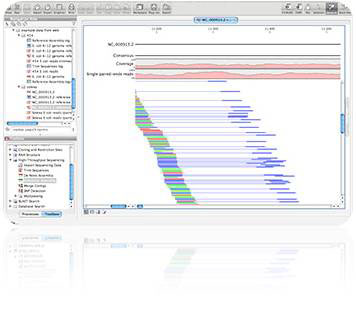 Illumina's HiSeq software automatically makes base calls and generates FASTQ sequence and quality files, which are delivered to you. As a separate service, the core's experienced bioinformatics team can perform custom analysis and alignments.
Illumina's HiSeq software automatically makes base calls and generates FASTQ sequence and quality files, which are delivered to you. As a separate service, the core's experienced bioinformatics team can perform custom analysis and alignments.
To discuss these options, please contact us.
Data Distribution
MGH users can directly access sequencing data through the Galaxy server.
To see instructions for retrieving data through Galaxy, click here.
External customers will receive summary and FASTQ data through secure FTP or other agreed upon data transfer mechanisms.
Data Retention
Sequence files are stored on an off-site server for 2 months. Too much data is generated to guarantee long-term storage longer than 2 months.
A Note About Turnaround Time
The size of the sequencing queue is highly variable and thus the wait time for sequencing your sample is often difficult to predict. Upon sample submission AND arrival, your samples are entered into a queue and run on the next available FULL flow cell that matches your run type. Thus, submitting more samples (e.g. an entire flowcell-worth) typically reduces wait time.
Once your samples have been assigned to a flowcell, the actual sequencing time is dependent upon the type of run and number of cycles, with typical run times shown below:
50 single read: ~2 days
50 paired-end: ~5 days
100 single read: ~4 days
100 paired-end:~8 days
SR50 and PE50 runs are most common, so samples submitted for these run types (if not part of a full flow cell) are generally run sooner than ones requesting SR100 and PE100.